

Highlight
Progress towards in-silico drug design
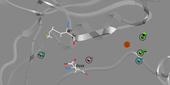
The pharmacophoric elements of the PERK binding site.
Achievement/Results
Prof. Timothy J. Cardozo and his team at NYU Sackler Institute have developed the first pharmacophore model for selective PERK inhibition as well as 14 selective PERK inhibitors using in silico drug design approaches along with experimental validation. This development could yield novel anti-cancer drug leads against a fundamental mechanism for tumor cell survival.
The unfolded protein response (UPR) is a critical survival mechanism for tumor cells. Chemotherapeutic drugs may induce cellular stress in cancer cells that the UPR helps overcome, suggesting that inhibition of the UPR in tumors would be a novel anti-cancer approach to target the very stress responses in cancer cells that hinder existing anti-tumor treatments. Among the three prominent UPR transducers, the protein kinase PERK has a broader range of cellular effects. Most importantly, its critical role in tumor progression has been demonstrated by the observation that compromising PERK function inhibits tumor growth in mice. Therefore, inhibiting the kinase activity of PERK would be an important and novel therapeutic intervention in cancer. To date, however, no specific small molecule inhibitor of PERK has been identified.
Prof. Timothy Cardozo and his team identified three structural determinants that are important in establishing selective PERK inhibition by small molecules. Compounds inhibiting PERK-mediated phosphorylation in an in vitro kinase inhibition assay were identified using molecular modeling, virtual library screening and chemoinformatics technologies. The most potent PERK selective compound utilizes three specific kinase active site contacts that, when lost in chemically similar compounds, abrogate the inhibition. This pharmacophore model could be used to advance the PERK targeted drug discovery effort, and may eventually lead to novel anti-cancer drug leads. A patent application based on this work is currently in the process.
Address Goals
It is generally recognized that drug discovery is a very time and resource consuming process. To find a drug against a disease protein is similar to searching for a needle in a haystack. Over the years, drug discovery has moved toward more rational strategies based on our increasing understanding of the fundamental principles of protein-ligand interactions. The role of computational models is to exploit structural and functional information in order to better understand specific molecular recognition events of the target macromolecule with candidate hits, ultimately leading to novel inhibitors and a pathway for optimization. Distinct from the traditional high throughput screening drug discovery approach, in this interdisciplinary project, Prof. Timothy Cardozo and his team employed a structure-based computational inhibitor design approach. This was combined with experimental validation to reveal the important protein-ligand atomic contacts responsible for selective PERK inhibition. This finding could accelerate PERK targeted drug discovery, a novel therapeutic intervention in cancer.
